
by Dr. Neil Taylor
Artificial intelligence is rapidly redefining what’s possible in ligand design. In this article, we explore recent breakthroughs in AI in drug discovery and computational chemistry, and how they are accelerating pharmaceutical innovation.
Just as AI/ML models like AlphaFold revolutionised structural biology by learning from experimental protein data, similar approaches are now reshaping how researchers identify and optimise new therapeutic compounds. At DesertSci, we are at the forefront of applying these innovations through proprietary technologies that bridge computational design and experimental feasibility.
For companies pursuing AI drug discovery, ligand design is undergoing a major transformation. Traditional methods are now being enhanced – or replaced – by AI models trained on vast repositories of experimental data. These techniques combine chemical similarity principles with the capability of advanced large language models (LLMs) to accelerate the discovery of novel compounds.
Modern computational approaches use different descriptors to analyse chemical structures:
Together, these methods allow researchers to rapidly assess and design new molecules with improved potency, selectivity, and drug-like properties.
Meanwhile, large language models for chemistry – trained on SMILES (Simplified Molecular Input Line Entry System) data – are opening new doors. These models “learn” the grammar of chemical structures, proposing novel compounds by predicting how atoms and bonds might arrange. Although still in early stages, they show tremendous promise in expanding chemical space, despite occasional challenges in accurately handling stereochemistry and bonding rules.
Another exciting advancement is chemical reaction prediction using AI. Modern tools can now:
These tools greatly enhance large scale virtual screening and compound library generation, helping projects move from concept to synthesis more swiftly than ever before.
One area where artificial intelligence in drug discovery and development shows immense promise is in designing inhibitors for KRAS G12D – a mutation commonly found in pancreatic, colorectal, and lung cancers.
KRAS is a gene that helps regulate cell division. When mutated at codon 12 (substituting glycine with aspartic acid – hence G12D), it can cause continuous signalling for cell growth, driving cancer progression. Designing molecules that can effectively target KRAS G12D has long been considered highly challenging.
Viper uses a reverse engineering approach:
This process enables the creation of diverse compound libraries, supporting both:
In a practical application targeting KRAS G12D, researchers developed a series of molecules featuring methyl-naphthalene substituents. Viper suggested novel modifications – such as ethyne-naphthalene variants – to optimise binding interactions. These candidates were ranked using DesertSci’s Scorpion platform, a network- and hotspot-based scoring system.
Through advanced computational chemistry, DesertSci’s algorithms:
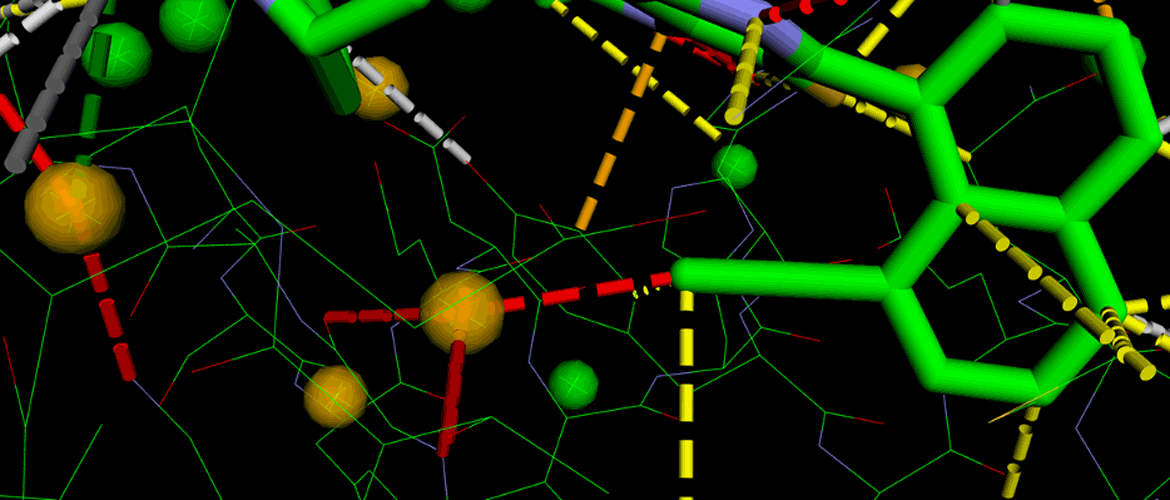
Figure 1: shows a close-up of pdb entry 8t4v, where Viper successfully pinpoints a favourable hydrogen bond between a ligand’s carbon atom and a networked water molecule – guiding scientists to an area where small molecular modifications can significantly boost binding affinity.
This case study exemplifies how AI in drug development can dramatically streamline lead optimisation.